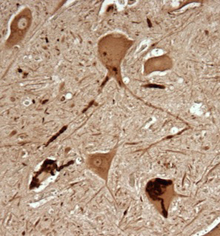
The SOD1 protein forms distinctive clumps in neurons affected by ALS, which can be detected with the C4F6 antibody.
This is spinal cord tissue from a patient with familial ALS.
Researchers at Emory University School of Medicine have demonstrated a way to detect the toxic form of a protein involved in some types of ALS (amyotrophic lateral sclerosis).
The scientists can see knots of the toxic protein within motor neurons, the type of cell killed as ALS progresses, by using an antibody to stain cells on microscope slides. Further study of the antibody could reveal more about how the toxic protein kills cells and why ALS specifically affects motor neurons.
The results were published in Proceedings of the National Academy of Sciences.
"It's a really exciting tool," says Terrell Brotherton, first author of the paper and a graduate student in Emory's Neuroscience program. "The antibody staining closely reflects the pathology of the disease, which is why it's so useful."
Brotherton is working in the laboratory of Jonathan Glass, MD, professor of neurology and director of the Emory ALS Center.
Antibodies are proteins produced by the immune system. Biomedical researchers often use "monoclonal" (homogenous) antibodies as tools in the laboratory, because each type of antibody sticks to something different.
The antibody Brotherton is studying reacts against a mutant form of the SOD1 (superoxide dismutase 1) protein. Most cases of ALS are sporadic, meaning that the causes are unknown. However, ten percent of ALS cases are inherited, and 20 percent of those are linked to mutations in the gene encoding SOD1, an antioxidant enzyme. Mice with a mutant SOD1 gene develop a disease that resembles ALS.
ALS kills motor neurons in the brain and spinal cord, causing the brain to lose ability to control muscles in the body. Over the course of a few years, progression of the disease leads to paralysis and problems with swallowing, eating, and breathing.
In spinal cord samples from patients with mutant SOD1, antibody staining reveals that some motor neurons have dark clumps, while others appear to lack them, even though they're next to others where the apparently toxic form of SOD1 is plentiful.
Brotherton has identified some examples of motor neurons that stain with the antibody and look like they're dying. In contrast, sensory neurons do not appear to contain the toxic protein.
"In patients who carry this mutation, mutant SOD protein is present in every cell in the body," she says. "The mystery is why only some motor neurons get sick."
The antibody (called C4F6) was generated by scientists at Laval University in Quebec. It detects clumps of SOD1 in spinal cord samples from animals that produce one mutant form of SOD1, and also human patients with a different mutation in the same gene. This means that the antibody probably recognizes something shared by the shapes adopted by the mutant proteins. However, the antibody does not appear to detect toxic protein in samples from patients with sporadic ALS, who have normal SOD1 in their cells, Brotherton says.
"It's not clear how C4F6 is able to distinguish toxic and innocuous protein," she says. "If we can figure out what exactly C4F6 reacts with, then perhaps we can figure out what the toxic form of the mutant protein is."
Recent research by other laboratories indicates that mutant SOD1 protein can grab other molecules of the same protein and clump together, resembling the prion particles that cause neurodegenerative diseases such as Creutzfeldt-Jakob disease. Once formed, these clumps may be able to grow and travel from cell to cell. The peculiar behavior of mutant SOD1 protein doesn't explain how all cases of ALS start and progress, but studying it could give more clues about how and why it kills certain cells.
The research was supported by the National Institute of Aging, the National Institute of Neurological Disorders and Stroke, and the Packard Center for ALS Research.
Reference: T.E. Brotherton, Y. Li, D. Cooper, M. Gearing, J. Julien, J.D. Rothstein, K. Boylan and J.D. Glass. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. PNAS Early Edition (2012).